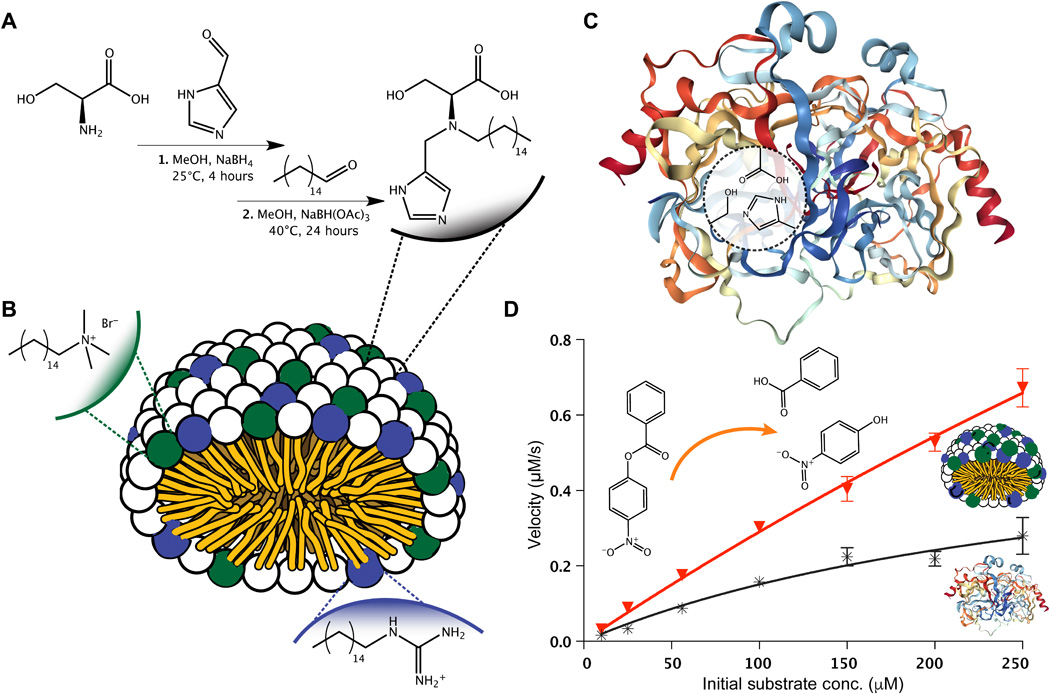
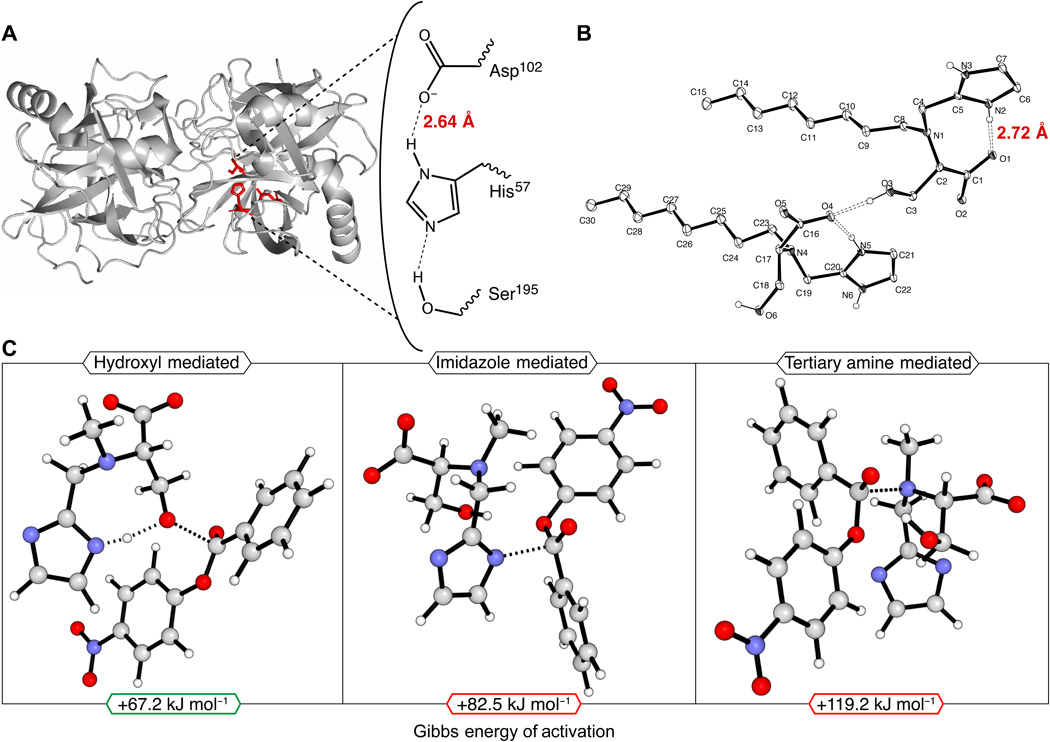
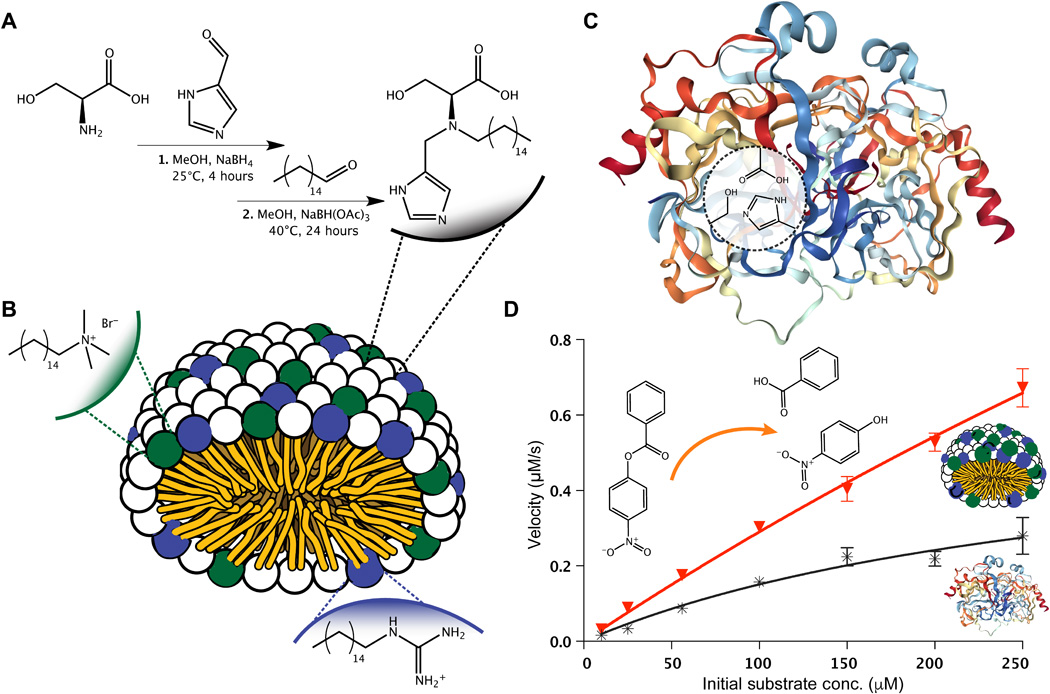
MATERIALS AND METHODS
General information
All commercially obtained solvents and reagents were used without further purification. Analytical thin-layer chromatography was carried out on Merck silica gel 60 F254 glass plates, and flash chromatography was performed on Merck silica gel 60 (70 to 230 mesh).
Visualization was accomplished with short-wave ultraviolet (UV) light and/or KMnO4 staining solution followed by gentle heating. Surfactants were purified by preparative reverse-phase high-performance liquid chromatography (RP-HPLC) on a Biotage SP1 HPFC Flash Purification System using a reverse-phase Biotage SNAP Cartridge (KP-C18-HS, 60 g). For synthesis characterization, 1H and 13C solution-state NMR were recorded on a Varian Unity Inova 500 (500 MHz for 1H and 125 MHz for 13C) or a Varian Unity Inova AS600 (600 MHz for 1H and 150 MHz for 13C) spectrometer. Chemical shifts δ are reported relative to the resonance signal of 1H or 13C cores of tetramethylsilane and in parts per million. The 1H spectra were calibrated by setting the solvent peaks, caused by remaining traces of protons, to values known from the literature (δCHCl3 = 7.26 ppm, δCD3OH = 4.87 ppm, and δD2O = 4.79 ppm). The coupling constants J are reported in hertz.
CMC measurements—Pyrene method
According to the literature (
60), a pyrene stock solution was prepared by dissolving pyrene (5 mg) in methanol (10 ml) and diluting 20-fold with methanol. Pyrene was stored under an inert atmosphere at 4°C, and stock solutions were prepared fresh each day. Surfactant stock solutions (~0.1 M) were prepared in dimethyl sulfoxide (DMSO) or borate (pH 9.0) (Na
2B
4O
7·10H
2O) buffer according to the individual surfactant solubilities. For the fluorescence assay, an appropriate volume of surfactant stock solution (surfactant final concentration range, 0.01 to 0.8 mM) was transferred to a quartz fluorescence cuvette. Pyrene stock solution (50 μl) and either deionized water or borate buffer (pH 9.0) were added to the cuvette for a final volume of 2.5 ml. In the cases where the surfactant was dissolved in DMSO, a small aliquot of DMSO was added to the cuvette to maintain a constant DMSO concentration for all surfactant concentrations [final DMSO concentration, <1% (v/v)]. The fluorescence emission spectrum of pyrene was measured immediately after mixing using an emission wavelength scanning mode from 360 to 400 nm over 90 s. The excitation wavelength was 334 nm (slit width, 8 nm), and emission slit width was 2 nm. The first and third fluorescent vibrational peaks (
I1 and
I3) were recorded at 373 and 384 nm, respectively, and the ratio was plotted against surfactant concentration for the CMC calculations. The midpoint of the inflection of
I1/
I3 versus surfactant concentration was taken as the CMC value in all cases (fig. S1).
Esterolysis assay with p-nitrophenyl benzoate (representative assay for the three-component catalyst)
Catalyst (ACT surfactant, 2.24 μM) was dissolved with cetyl ammonium bromide (CTAB; 0.8 mM) and hexadecylguanidinium chloride (Gu-C16; 0.3 mM) in a borate (Na2B4O7·10H2O) buffer at pH 9 (cbuffer = 100 mM) with vigorous stirring. Control reactions lacking either CTAB, ACT surfactant, or Gu-C16 were also conducted with the same concentrations. A stock solution of substrate p-nitrophenyl benzoate was made up in acetonitrile and added to the buffered solution containing surfactants (csubstrate,initial = 10, 25, 56, 100, 150, and 250 μM). A small aliquot of acetonitrile was added to the reference cell to maintain consistency [acetonitrile concentration, <5% (v/v)]. Following mixing, the reaction was immediately monitored via UV light absorption at 405 nm at room temperature in a Cary 60 UV-visible spectrophotometer (Agilent). The kinetics of ester substrate consumption was calculated via nonlinear regression using the plotting software GraphPad Prism 8, under an assumption of Michaelis-Menten kinetics. The background hydrolysis rate without the addition of catalyst or surfactants was determined using a first-order rate assumption, where kcat is equal to the khydrolysis for the reaction.
For the enzyme-catalyzed control reaction, α-chymotrypsin (from bovine pancreas type II, lyophilized powder, Sigma-Aldrich) (2.2 μM) was dissolved as received in borate (Na2B4O7·10H2O) buffer at pH 9 (cbuffer = 100 mM), containing CTAB (0.8 mM). A stock solution of substrate p-nitrophenyl benzoate was made up in acetonitrile and added to the buffered solution containing surfactants (csubstrate,initial = 10, 25, 56, 100, 150, and 250 μM), as per the surfactant experiments above. The hydrolysis reaction was assessed over 10 min and compared directly with the results of the cosurfactant systems.
X-ray crystallography of ACT-C8 surfactant
The crystal data of the ACT-C8 surfactant were collected on a charge-coupled device diffractometer using Cu-Kα radiation (graphite crystal monochromator = 1.54184 Å). The structure was solved by direct methods (SHELXT) and difference Fourier synthesis. Thermal ellipsoid plots were generated using the program ORTEP-3 integrated within the WinGX suite of programs. C15H27N3O3: M = 297.39, T = 130.0(2) K, λ = 1.54184 Å; monoclinic, space group P21: a = 12.9624(5), b = 8.4191(3), c = 14.9957(6) Å, β = 93.510(3), V = 1633.44(11) Å3, Z = 4, Z′ = 2, Dc = 1.209 Mg M−3 μ(Cu-Kα) = 0.685 mm−1, F(000) = 648; crystal size, 0.65 mm by 0.21 mm by 0.03 mm. θmax = 76.48, 9264 reflections measured, 4774 independent reflections (Rint = 0.058), the final R = 0.0567 [I > 2σ(I), 4459 data], and wR(F2) = 0.1559 (all data) Goodness-of-fit = 1.073, absolute structure parameter 0.0(2). Cambridge Crystallographic Data Centre code: 1900732.
NMR of ACT-C8 surfactant
NMR samples were prepared by either dissolving ACT-C8 in buffer [50 mM borate (pH 9.0), 0.05 mM DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid), 10% (v/v) D2O] or in 75 mM deuterated CTAB [50 mM borate (pH 9.0), 0.05 mM DSS, 10% (v/v) D2O] to reach a final compound concentration of ca. 5 mM. NMR spectra were obtained at 298 K on an 800-MHz Bruker Avance II equipped with a TCI CryoProbe. Chemical shifts were referenced to DSS at 0 ppm. Data were processed in TopSpin (Bruker) and analyzed using the CCPNmr Analysis program (Laue 2005 Proteins 59, 687). 1H homonuclear correlation spectroscopy, TOCSY (mixing time τmix = 100 ms), and NOESY (τmix = 100 and 500 ms) were run using 4000 and 512 points in the direct and indirect dimensions, respectively.
Synthetic procedures
Reductive amination of l-serine with 1H-imidazole-2-carboxaldehyde (preparation of ACT molecule)
l-Serine (1.05 g, 10 mmol, 1.00 eq) and sodium hydroxide (420 mg, 10.5 mmol, 1.05 eq) were dissolved in methanol (cL-serine ≈ 85 mM), and 1H-imidazole-2-carboxaldehyde or 1H-imidazole-4-carboxaldehyde (1.06 g, 11 mmol, 1.10 eq) was added to the solution with stirring. The mixture was stirred at 40°C for 30 min and afterward allowed to cool down to room temperature over 4 hours while stirring. Sodium borohydride (605.3 mg, 16 mmol, 1.60 eq) was added slowly, and the reaction mixture was stirred at room temperature for 30 min. Glacial acetic acid was added dropwise until the mixture reached approximately pH 4 to 5, and the resulting suspension was stirred at room temperature for a further 10 min. The product was obtained as the precipitate by filtration and washing with methanol. ACT was collected as a fine, white solid (yield 99%).
[ACT (2-imidazole isomer)] 1H NMR (600 MHz; D2O, 298 K): δ = 7.23 (s, 2H, aromatic), 4.31 (d, 2J = 15.1 Hz, 1H, CHHN), 4.23 (d, 2J = 15.1 Hz, 1H, CHHN), 3.95 to 3.77 (m, 2H, CH2OH), 3.56 (dd, 3J = 5.3 Hz, 3J = 4.1 Hz, 1H, CHCO2−). MS [electrospray ionization (ESI)] was calculated for C7H11N3O3H+ ([M + H]+): 186.09. Found: 186.09.
[ACT (4-imidazole isomer)] 1H NMR (400 MHz, CD3OD, 298 K): δ = 7.66 (s, 1H, Im), 7.17 (s, 1H, Im), 4.15 (d, J = 13.9 Hz, 1H, CH2OH), 4.11 (d, J = 13.9 Hz, 1H, CH2OH), 3.84 (dd, J = 12.9, 3.9 Hz, 1H, CH2Im), 3.78 (dd, J = 12.9, 3.9 Hz, 1H, CH2Im), 3.57(t, J = 7.4, 3.8 Hz, 1H, CHCH2OH). MS (ESI) was calculated for C7H11N3O3H+ ([M + H]+): 186.09. Found: 186.09.
Preparation of n-octan-1-al from n-octan-1-ol
According to the literature (
61),
n-Octan-1-ol (1.3 g, 10 mmol, 1.0 eq) was dissolved in dichloromethane (
calcohol ≈ 65 mM), and pyridinium dichromate (4.5 g, 12 mmol, 1.2 eq) was added to the solution portionwise. The mixture was stirred at room temperature for 8 hours. The suspension was filtered through filter paper and a short silica pad. The solid residue was washed several times with diethyl ether. The solvents were removed in vacuo, and the crude product was purified by flash chromatography (hexane/ethyl acetate = 19:1) to yield a waxy colorless solid (819 mg, 64%).
1H NMR (400 MHz, CDCl
3, 298 K): δ = 9.74 (t,
J = 1.9 Hz, 1H, C
HO), 2.40 (td,
J = 7.4, 1.9 Hz, 2H, C
H2CHO), 1.60 (m, CH
2, 2H), 1.35 to 1.19 (m, CH
2, 8H), 0.86 (t,
J = 6.8 Hz, 3H).
Preparation of n-hexadecan-1-al from n-hexadecan-1-ol
1-Hexadecanol (5.00 g, 20.6 mmol, 1.00 eq) was dissolved with stirring in dichloromethane (150 ml). Pyridinium dichromate (9.40 g, 25.0 mmol, 1.20 eq) was added portionwise, and the mixture was stirred for a further 5 hours. The suspension was filtered over a short silica pad and washed several times with dichloromethane and ethyl acetate, and the filtrate was dried in vacuo. Purification of the crude product by column chromatography (hexane/ethyl acetate = 19:1) yielded target aldehyde as a colorless solid (3.69 g, 15.3 mmol, 74%). 1H NMR (500 MHz, CDCl3, 298 K): δ = 9.76 (t, 3J = 1.9 Hz, 1H, CHO), 2.41 (td, 3J = 7.4 Hz, 3J = 1.9 Hz, 2H, CH2CHO), 1.66 to 1.58 (m, 2H, CH2), 1.35 to 1.21 (m, 24H, CH2), 0.88 (t, 3J = 6.9 Hz, 3H, CH3).
Reductive amination of ACT with n-octan-1-al (preparation of ACT-C8 surfactant)
The corresponding ACT (1.00 eq) and sodium hydroxide (1.05 eq) were dissolved in methanol (cprecursor ≈ 100 mM). n-Octan-1-al (1.20 eq) and sodium triacetoxyborohydride (1.40 eq) were added to the solution, and the mixture was stirred at 40°C for 2 hours. Another two batches of n-octan-1-al (2 × 1.20 eq) and sodium triacetoxyborohydride (2 × 1.40 eq) were added to the solution every 2 hours, and after 4 hours, the mixture was left to stir at 40°C for a further 20 hours. The reaction was quenched with glacial acetic acid, and the solvents were removed in vacuo. The crude product was washed with ethyl acetate and purified by RP-HPLC with an H2O/acetonitrile gradient (19:1→1:19). (Both derivatives a pale-yellow solid, 2-isomer = 60%, 4-isomer = 84%.) (ACT-C8 2-isomer) 1H NMR (500 MHz; D2O, 298 K): δ = 7.78 (s, 1H, Im), 7.32 (s, 1H, Im), 4.34 to 4.18 (m, 2H, ImCH2N), 4.00 to 3.82 (m, 2H, CH2OH), 3.57 (dd, 3J = 6.9 Hz, 3J = 5.2 Hz, 1H, CHCO2H), 2.86 to 2.70 (m, 2H, CH2CH2N), 1.44 to 1.30 (m, 2H, CH2CH2N), 1.24 to 1.03 (m, 10H, CH2), 0.75 (t, 3J = 6.8 Hz, 3H, CH3). MS (ESI) was calculated for C15H27N3O3H+ ([M + H]+): 298.21. Found: 298.24.
Reductive amination of ACT with n-hexadecan-1-al (preparation of ACT-C16 surfactant)
The corresponding ACT (185 mg, 1.0 mmol, 1.00 eq) was dissolved in methanol and glacial acetic acid (camino acid precursor ≈ 12.3 M) with stirring at 60°C. C16 aldehyde (360 mg, 1.5 mmol, 1.50 eq) was added, and the reaction was mixed for 20 min. Sodium triacetoxyborohydride (420 mg, 2.0 mmol, 2.0 eq) was added carefully to the solution, and stirring was continued at 60°C for 4 hours. Another batch of aldehyde (360 mg, 1.5 mmol, 1.50 eq) and Sodium triacetoxyborohydride (420 mg, 2.0 mmol, 2.0 eq) was added, and the mixture was stirred at 60°C for a further 24 hours. The reaction was quenched with water, and the solvents were removed in vacuo at elevated temperature. The crude product was extracted with methanol and washed several times with ethyl acetate before being purified by reverse-phase column chromatography (H2O/methanol = 19:1→1:19→0:1) to yield ACT-C16 surfactant (213 mg, 0.52 mmol, 52%). 1H NMR (400 MHz, CD3OD, 298 K): δ = 7.77 (s, 1H, Im); 7.35 (s, 1H, Im); 4.51 (d, J = 13.9 Hz, 1H, CH2OH); 4.42 (d, J = 13.9 Hz, 1H, CH2OH), 4.17 (dd, J = 12.9, 3.9 Hz, 1H, CH2Im), 4.05 (dd, J = 12.9, 3.9 Hz, 1H, CH2Im); 3.82 (dd, J = 7.4, 3.8 Hz, 1H, CHCH2OH); 2.24 (t, J = 7.4 Hz, 1H, CH2N); 1.57 (p, J = 7.2 Hz, 2H, CH2CH2N); 1.40 to 1.22 (m, 26H, CH2); 0.88 (t, J = 7.2 Hz, 3H, CH3). MS (ESI) was calculated for C23H43N3O3H+ ([M + H]+): 410.33. Found: 410.34.
1-Hexadecylguanidinium chloride (Gu-C16)
According to the literature (
43), 1-hexadecylamine (723 mg, 3.0 mmol, 1.0 eq) was dissolved in methanol (15 ml) at 40°C with stirring. 1H-pyrazole-1-carboxamidine hydrochloride (438 mg, 3.0 mmol, 1.0 eq) was added, and stirring was continued at 40°C for 3 days. Solvent was removed in vacuo, and the crude product was washed three times with hot acetone to yield target surfactant
Gu-C16 as a colorless solid (850 mg, 2.9 mmol, 97%).
1H NMR (400 MHz, CD
3OD, 298 K): δ = 3.14 (
t,
J = 7.1 Hz, 2H), 1.56 (
p,
J = 7.2 Hz, 2H), 1.41 to 1.22 (
m, 26H), 0.87 (
m, 3H). MS (ESI) was calculated for C
17H
33N
3H
+ ([M + H]
+): 284.30. Found: 284.32.
Preparation of (OH–COOH) control structure [(S)-3-hydroxy-2-(octylamino)propanoic acid]
l-serine (210 mg, 2.0 mmol, 1.0 eq) was suspended in 15 ml of methanol at 50°C, and sodium hydroxide (88 mg, 1.4 mmol, 1.1 eq) was added with stirring. 1-Octanal (310 mg, 2.4 mmol, 1.2 eq) was then added, and the reaction was mixed overnight at room temperature. NaBH4 (150 mg, 4.0 mmol, 2.0 eq) was then added, and the reaction mixture was stirred for a further 1 hour at room temperature. Concentrated hydrochloric acid (32%, ~0.7 ml) was added to quench the reaction mixture, the suspension was filtered, and solvent was removed in vacuo. The crude product was washed with ethyl acetate and then purified via reverse-phase column chromatography (MeOH/H2O 19:1→0:1) to yield target structure (239 mg, 1.1 mmol, 54%). 1H NMR (400 MHz; CD3OD, 298 K): δ = 3.93 (dd, J = 11.8, 4.0 Hz, 2H, CH2OH), 3.82 (dd, J = 11.8, 6.4 Hz, 2H, CH2OH), 3.46 (dd, J = 6.4, 4.0 Hz, 1H, CHCH2OH), 2.95 (m, 2H, CH2NH), 1.67 (p, J = 7.6 Hz, 2H), 1.38 to 1.13 (m, 10H, CH2), 0.97 to 0.80 (m, 3H, CH3). MS (ESI) was calculated for C11H23NO3H+ ([M + H]+): 218.17. Found: 218.19.
Preparation of (Im–COOH) control structure [(R)-3-(1H-imidazol-5-yl)-2-(octylamino)propanoic acid]
l-histidine (620.8 mg, 4.0 mmol, 1.0 eq) was suspended in 20 ml of methanol at 50°C, and sodium hydroxide (168 mg, 4.2 mmol, 1.1 eq) was added with stirring. 1-Octanal (620 mg, 4.8 mmol, 1.2 eq) was then added, and the reaction was mixed overnight at room temperature. NaBH4 (300 mg, 8.0 mmol, 2.0 eq) was then added, and the reaction mixture was stirred for a further 1 hour at room temperature. Concentrated hydrochloric acid (32%, ~1.0 ml) was added to quench the reaction mixture, the suspension was filtered, and the solvent was removed in vacuo. The crude product was washed with ethyl acetate and a small volume of water and then purified via reverse-phase column chromatography (MeOH/H2O 19:1→0:1) to yield target structure (639 mg, 2.4 mmol, 60%). 1H NMR (400 MHz; CD3OD, 298 K): δ = 8.74 (s, 1H, Im); 7.38 (s, 1H, Im); 3.71 to 3.60 (m, 1H, CHCOOH), 3.54 to 3.43 (m, 2H, CH2Im), 3.08 to 2.97 (m, 2H, CH2NH), 1.47 to 1.35 (m, 2H, CH2CH2NH), 1.31 to 1.07 (m, 10H, CH2); 0.79 to 0.7 (m, 3H, CH3). MS (ESI) was calculated for C14H25N3O2H+ ([M + H]+): 268.20. Found: 268.21.
Preparation of (Im–OH) control structure [(R)-3-(1H-imidazol-5-yl)-2-(octylamino)propan-1-ol]
l-histidinol dihydrochloride (430 mg, 2.0 mmol, 1.0 eq) was suspended in 15 ml of methanol at 50°C, and sodium hydroxide (180 mg, 4.2 mmol, 2.1 eq) was added with stirring. The cloudy suspension was filtered, 1-octanal (260 mg, 2.1 mmol, 1.05 eq) was then added, and the reaction was mixed overnight at room temperature. NaBH4 (90 mg, 2.4 mmol, 1.2 eq) was then added, and the reaction mixture was stirred for a further 1 hour at room temperature. Concentrated hydrochloric acid (~0.6 ml) was added to quench the reaction mixture, the suspension was filtered, and the solvent was removed in vacuo. The crude product was washed several times with ethyl acetate, extracted into methanol, and purified via reverse-phase column chromatography (MeOH/H2O 19:1→0:1) to yield target structure (214 mg, 0.8 mmol, 42%). 1H NMR (400 MHz; CD3OD, 298 K): δ = 8.81 (s, 1H, Im); 7.43 (s, 1H, Im); 4.40 to 4.34 (m, 2H, CH2OH), 4.18 to 4.07 (m, 2H, CH2Im); 3.42 (dd, J = 7.4, 3.8 Hz, 1H, CHCH2OH); 3.11 to 2.97 (m, 2H, CH2CH2N); 2.23 to 2.06 (m, 2H, CH2CH2N); 1.39 to 1.10 (m, 26H, CH2); 0.80 (t, J = 7.2 Hz, 3H, CH3). C14H26N3O− ([M − H]−): 252.22. Found: 252.21.
Computational procedures
Quantum mechanics
All electronic structure calculations were performed with the Gaussian 16, Revision A.03 (
62) software package. All density functional theory calculations were performed using the M06-2X functional (
63) in conjunction with the 6-31+G(d,p) basis set. Implicit solvent effects for all calculations performed were obtained using the SMD (solvent model density) (
64) solvation model for water. The ACT molecules were truncated by replacement of the extended alkyl chains with a single methyl group and conformationally searched to locate the global minimum energy structure; guanidine was also truncated by replacement of the extended alkyl chain with a single methyl group, so as to determine the possible role of the positively charged headgroup. All entropies, zero-point vibrational energies, and thermal corrections were scaled by recommended scale factors (
65); vibrational analysis was performed on all geometries to confirm (i) the absence of imaginary frequencies for minimum energy structures and (ii) the presence of a single imaginary frequency for transition-state structures.
Molecular dynamics
MD simulations were performed using the GROMACS 2018.1 engine with parameters from the GROMOS54a7 force field (
66,
67). All-atom parameter sets for each surfactant (CTAB, molid 10929; Gu-C16, molid 306970), uncharged catalyst (ACT-C8, molid 306428; ACT-C16, molid 306429), charged catalyst (ACT-C8, molid 306968; ACT-C16, 306969), and substrate (PNB, molid 9503) corresponding to the predicted ionization states at pH 9 were obtained using the Automated Topology Builder V.2.2 (
68). To remove any conformational bias from the system setup, triplicate systems were prepared in which molecules of substrate, catalyst, and surfactant were randomly orientated in a cubic dodecahedron box at the relative concentrations used in experimental assays (table S2) and solvated with a 7-nm buffer of explicit simple point-charge water. The concentration of surfactant species was increased 10-fold to facilitate sampling. Ions were added as required to neutralize overall system charge. The initial configuration was relaxed using standard steepest descent minimization using at least 1000 steps before being equilibrated for 10 ns in the isothermal–isobaric (
NpT) ensemble to stabilize the system density. Each catalytic system (simulations 1 to 20; table S2) was allowed to self-assemble for 100 ns under
NpT. Single CTAB controls (simulations 21 to 25; table S2) were run at various concentrations under the same conditions for 300 ns each. Periodic boundary conditions were used, and long-range electrostatics were calculated using the particle-mesh Ewald method with a cutoff of 14 Å (
69). The temperature in all simulations was set to 300 K and controlled via the v-rescale thermostat (
70); the initial velocities of all particles were randomly generated. Pressure coupling was handled with the Berendsen barostat during equilibration and with the Parrinello-Rahman barostat for dynamics. The LINCS (Linear Constraint Solver) algorithm (
71) was used to constrain bonds in conjunction with an integration time step of 2 fs. All trajectory images were produced in VMD (visual molecular dynamics) (
72). All data were visualized using Python.
Aggregation numbers and component ratios
Micelle aggregation numbers and component ratios were calculated using a Python program using MDTraj (
73) written for this purpose that recursively considers the neighbors of each micelle component within a given cutoff to define an aggregate, until no more neighbors are found. A 3-Å cutoff was used, and all micelle components in the simulation box were found using this scheme. Micelle aggregation numbers reported (figs. S4 and S5) are for a single frame of each replicate after 100 ns, after periodicity was considered. Component ratios are reported as the sample mean across replicates at 100 ns, after combining data from simulations run at different protonation states, in fig. S6.
Hydration number
Hydration numbers were calculated by searching for solvent molecules near a given atom within a cutoff of 4 Å, which was determined to be optimal by generating radial distribution functions (fig. S10) for the solvent surrounding each atom of interest from simulations containing only the catalyst in water. Data reported are the distribution for each simulation over the whole simulation time, as reported in table S2. The hydration numbers of catalytically relevant atoms of interest are given in figs. S7 to S9.
Intramolecular distances
Intramolecular distances between relevant atoms of the catalysts were measured as the Cartesian distance between the groups at each frame of the final trajectory, after considering periodic boundary conditions. The intramolecular distances of each catalyst monomer were considered separately, and the overall distance data reported are the distributions over the total simulation time of each set of replicates (figs. S11 and S12).